
Carol Rees Parrish, M.S., R.D., Series Editor
22 PRACTICAL GASTROENTEROLOGY • APRIL 2017
NUTRITION ISSUES IN GASTROENTEROLOGY, SERIES #162
John K. DiBaise
Protein Losing Enteropathy:
Diagnosis and Management
Andrew P. Copland, MD
1
John K. DiBaise, MD
2
1
Division of Gastroenterology and Hepatology,
University of Virginia Health System, Charlottesville,
VA
2
Division of Gastroenterology and Hepatology,
Mayo Clinic in Arizona, Scottsdale, AZ
Andrew P. Copland
INTRODUCTION
P
rotein-losing enteropathy (PLE), sometimes
referred to as protein-losing gastroenteropathy,
is an unusual cause of hypoproteinemia and is
characterized by the shedding of large quantities of
protein from the gastrointestinal (GI) mucosa. PLE may
result from a wide variety of etiologies and can be both
a diagnostic and therapeutic challenge to the practicing
gastroenterologist. The clinical presentation of PLE
may also be complicated by micronutrient deciencies
related to the underlying etiology of the PLE. In some
cases, we have noted signicant vitamin deciencies
and deciency of essential fatty acids complicating
the care of these patients. Through the use of a case
illustration, we will explore a practical approach to the
evaluation and management of PLE.
In early 2016, a 51 year-old woman presented to the
GI clinic upon referral by a hematologist because of the
development of progressive hypoalbuminemia (albumin
2.6 g/dL) which had been identied approximately 1
year earlier.
She described one normal appearing stool
per day, denied any GI complaints, and her physical
examination was entirely normal.
PLE is generally considered to be a rare condition;
however, given a lack of systematic screening and a wide
variety of causes of hypoalbuminemia, its prevalence is
poorly understood. There are robust data describing an
incidence of up to 18% among survivors of the Fontan
procedure, used as treatment of the univentricular
congenital heart; however, data are much more limited
Protein losing enteropathy (PLE) is an uncommon etiology of hypoproteinemia. It is caused by protein
loss from compromised gastrointestinal (GI) mucosa as a result of GI mucosal diseases, GI tract
infections, as well as from disruptions of venous and lymphatic outow. The prevalence of PLE is
poorly understood given the wide variety of causes of both hypoalbuminemia and PLE, and due to a
lack of systematic screening. The evaluation of a potential PLE patient includes a careful assessment
for alternative causes of hypoalbuminemia and a measurement of GI tract protein loss. This review
provides the clinician with diagnostic criteria, as well as management and nutrition support options.
(continued on page 24)

24 PRACTICAL GASTROENTEROLOGY • APRIL 2017
NUTRITION ISSUES IN GASTROENTEROLOGY, SERIES #162
Protein Losing Enteropathy: Diagnosis and Management
for other causes of PLE.
1
A 2-3% prevalence of PLE
has been reported among Asian patients with systemic
lupus erythematosus (SLE).
2
In a study of 24 patients
with ileal Crohn’s disease in clinical remission, all
had laboratory evidence of PLE (although none had
clinical signs), suggesting that the prevalence of PLE
may be signicantly underrecognized.
3
Similarly, in
a study from 1975, 22% of 55 patients with primary
lymphedema who were screened for PLE were found
to have evidence of protein wasting from the GI tract.
4
Despite the poor understanding of its prevalence,
PLE should be a consideration in the evaluation of patients
who present with moderate to severe hypoalbuminemia
(serum albumin <3.0 g/dL), particularly those who
present with edema. Although some patients with PLE
present with severe GI symptoms such as diarrhea,
which can take on a secretory character, it is important
to recognize that not all patients suffering from PLE
will exhibit overt GI symptoms. In fact, the key clinical
characteristic of PLE is symptomatic hypoalbuminemia
which manifests most commonly as edema. Other
clinical manifestations generally reect the underlying
disease responsible for PLE.
Pathophysiology
The protein loss in the bowel typically results in serum
albumin levels <3.0 g/dL, and frequently <2.0 g/dL. In
the normal GI tract, only 1-2% of total daily protein is
lost through active intestinal secretions and mucosal
turnover.
5
This is signicantly different from the
dramatic protein losses from the GI tract seen in PLE,
which can result in daily loss of as much as 60% of
the total serum protein.
6
Because albumin contributes
about 80% of the total colloidal osmotic effect of human
serum due to its oncotic effect and afnity for sodium
ions, loss of serum albumin results in third-spacing of
uid and generally manifests clinically as peripheral
edema, ascites, and pleural effusions.
7
In addition to
symptomatic hypoalbuminemia, patients presenting
with PLE may be at increased risk of infection and
thrombosis due to concomitant stool loss of serum
immunoglobulins and key anticoagulant proteins
respectively, although neither occurs commonly.
In the context of increased serum protein loss, the
body will attempt to compensate by increasing protein
synthesis. As such, serum levels of rapid turnover
proteins including prealbumin, immunoglobulin E
(IgE), and insulin may remain normal.
8
In contrast,
(continued from page 22)
insufcient compensatory protein production and low
serum level is more often seen with slower turnover
proteins such as albumin, ceruloplasmin, brinogen,
transferrin, and immunoglobulins (other than IgE), as
the body has a less robust capacity to increase daily
production.
5
Albumin in particular is a slow turnover
protein with a half-life of about 25 days; there is also
evidence that the liver is unable to fully compensate
for sustained albumin losses.
7
Decreased serum levels
of lipids and trace elements have also been reported in
PLE, as has the presence of lymphopenia, particularly
in the setting of lymphatic obstruction or malnutrition.
The reduction of serum proteins other than albumin
seldom causes clinically signicant problems.
Her past medical history was notable for remote
peptic ulcer disease, hyperlipidemia, seasonal allergies
and persistent unexplained peripheral eosinophilia rst
discovered in 2010. In 1989, she underwent a vagotomy
and pyloroplasty for gastric outlet obstruction due
to peptic ulcer disease. Extensive evaluations of the
eosinophilia by specialists in infectious diseases,
hematology, allergy, immunology, and rheumatology
were unsuccessful in identifying an etiology.
Alternative causes of hypoalbuminemia
Other causes of hypoalbuminemia are diverse
and warrant careful thought when evaluating the
hypoalbuminemic patient. In particular, uid overload
(e.g., congestive heart failure), reduced protein synthesis
(e.g., chronic liver disease), and other sources of serum
protein losses (e.g., nephrotic syndrome) are important
Table 1. Causes of Hypoalbuminemia Other Than PLE
¨ Impaired Synthesis
o Chronic liver disease
¨ Increased Loss
o Nephrotic syndrome
¨ Dilution
o Volume overload in context of heart
failure
¨ Inflammation
o Acute inflammatory response (nega-
tive phase reactant)
o Chronic inflammatory response
NUTRITION ISSUES IN GASTROENTEROLOGY, SERIES #162
PRACTICAL GASTROENTEROLOGY • APRIL 2017 25
Protein Losing Enteropathy: Diagnosis and Management
has demonstrated superior sensitivity and negative
predictive value compared to A1AT clearance for the
diagnosis of PLE and has the added benet of not
requiring a prolonged stool collection.
12
These tests
may also be used to monitor response to treatment.
Upper endoscopy was subsequently performed
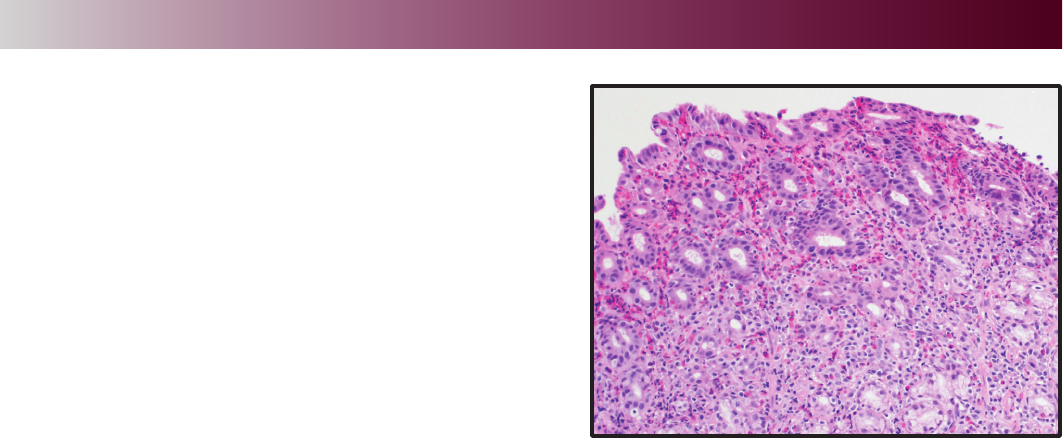
and was notable for patchy gastric erythema with an
atrophic appearance to the stomach. Biopsies from the
second portion of the duodenum demonstrated patchy
eosinophilia while biopsies from the duodenal bulb were
normal. Random biopsies from the stomach showed
marked eosinophilia without other abnormalities
(Figure 1). Based on the peripheral eosinophilia and
presence of eosinophils on the biopsy, the patient
was suspected to have eosinophilic gastroenteritis.
Interestingly, in 2010, she had undergone upper
endoscopy and colonoscopy to evaluate iron deciency
anemia. While both examinations were grossly
normal, random biopsies from the stomach revealed
a similar intense eosinophilic inammatory inltrate
throughout the mucosa and submucosa. Biopsies from
the duodenum, terminal ileum and colon were normal.
Evaluating Confirmed PLE
When a diagnosis of PLE has been determined,
additional testing is necessary in order to identify the
underlying cause and help direct treatment. PLE is
associated with a diverse set of diseases often affecting
multiple organ systems and can be divided into GI and
non-GI causes (Table 3). GI sources can be further
divided into erosive and nonerosive diseases of the
bowel that result in protein loss across the mucosal
membrane of the intestine and are detailed in Table 3.
to consider. This evaluation should include a careful
history and physical examination, as well as standard
evaluations of other causes of hypoalbuminemia noted
in Table 1.
A diagnostic evaluation revealed no evidence of
chronic liver disease, nephrotic syndrome or congestive
heart failure. Alpha-1-antritrypsin clearance was
found to be 341 mL/24 hours (normal, < 27 mL/24
hr), consistent with PLE.
Testing to Confirm a Diagnosis of PLE
The primary diagnostic test for PLE is stool testing
for the presence of alpha-1-antritrypsin (A1AT) (Table
2). A1AT is a protein that suffers minimal degradation
or active secretion in the GI tract and is of similar
molecular weight as albumin. By measuring A1AT
levels in both serum and a 24-hour stool collection,
A1AT clearance can be calculated as follows:
A1AT clearance = [(mL Stool) x (stool A1AT mg/
dL)] / [serum A1AT mg/dL]
An elevated A1AT clearance >27 mL/day reects
a general state of GI protein loss and has a sensitivity
of approximately 80%.
7
Diarrhea from any cause,
however, results in some obligate A1AT loss and, thus,
a higher threshold (>56 mL/day) may be required for
the diagnosis of PLE in this situation.
9,10
A1AT is also
sensitive to degradation by acid so, in the setting of a
hypersecretory state, this test is optimally performed
while the patient is receiving acid suppression.
11
Finally, A1AT testing of a spot stool specimen may
also show elevated levels in PLE, but this is a less
sensitive approach and is not recommended in the
initial diagnosis.
10
Use of a random stool A1AT level
coupled with serum A1AT level, however, may serve
as a convenient surveillance method for patients with
known PLE undergoing treatment or in remission.
There are a number of other methods to search
for protein loss in the GI tract, albeit none as widely
available or as safe as the A1AT clearance. Historically,
the gold standard test for PLE has been the fecal
excretion of
51
Cr labelled albumin, which requires
collection of stool for a minimum of 4 days.
7
It is not
only challenging for patients to complete a 4-day stool
collection but it exposes them to radiation and it is not
widely available. The
51
Cr-albumin clearance may be
useful when there is a high clinical suspicion in the
context of a negative A1AT clearance given its higher
sensitivity. An alternative is technetium 99m-labelled
human serum albumin (HSA) scintigraphy. This test
(continued on page 28)
Figure 1.

28 PRACTICAL GASTROENTEROLOGY • APRIL 2017
NUTRITION ISSUES IN GASTROENTEROLOGY, SERIES #162
Protein Losing Enteropathy: Diagnosis and Management
NUTRITION ISSUES IN GASTROENTEROLOGY, SERIES #162
Circulatory dysfunction from cardiac pathology
such as congestive heart failure (CHF), constrictive
pericarditis, and congenital heart disease can lead to
PLE. The most common cardiac cause of PLE occurs in
adults with congenital heart disease, a functional single
ventricle, treated as a child with a palliative Fontan
operation.
1
Post-Fontan patients make up the largest
cohort of patients with PLE described in the literature.
PLE is associated with a signicant morbidity and
mortality depending upon the underlying cause. The
ve-year mortality after diagnosis of PLE in the setting
of a Fontan procedure approaches 50%; however, recent
data suggest that advances in our understanding of the
disease may have improved this rather dismal outlook.
13
While data on morbidity and mortality associated
with PLE related to other causes are more limited,
malnutrition, volume overload, thrombophilia, and
secondary immunodeciency, likely have a signicant
impact on long-term outcomes.
Because the management of PLE is closely
tied to treating the underlying disease, when PLE is
identied, a thorough evaluation should be undertaken
to better characterize the state of the GI tract mucosa,
lymphatic system, and cardiovascular system. This
is best approached through upper and lower GI
endoscopy with mucosal biopsies as well as infectious
studies (focusing on chronic intestinal infections). If
conventional endoscopy does not yield a diagnosis,
video capsule endoscopy or small bowel enteroscopy
have been shown to be useful in patients with known
PLE.
14
Cross-sectional imaging of the abdomen and
pelvis, echocardiogram, lymphatic/hematologic tests
and, sometimes, diagnostic laparoscopy may also be
useful depending on the clinical presentation.
Mechanisms Causing PLE
As our understanding of PLE has improved, it has
become increasingly clear that a common link between
the various etiologies of the disease involves injury to,
or breakdown of, the GI epithelium causing increased
permeability. Conceptually, this pathology is clear
when considering PLE caused by mucosal diseases
such as inammatory bowel disease, eosinophilic
gastroenteritis, and microscopic colitis, for example.
Poor lymphatic drainage from congenital defects or
from signicant lymphatic obstruction may cause
loss of lymphatic uid into the GI tract through
direct hydrostatic forces.
15
Although the mechanism
responsible for PLE in systemic autoimmune disease
is unclear, it has been hypothesized that it results from
mucosal or capillary inammation caused by:
1.
local vascular injury mediated by complement
or vasculitis
2.
lymphatic damage through mesenteric
inammation, or
3.
increased endothelial permeability through
the effect of inammatory cytokines.
16
The cause of PLE associated with cardiovascular
diseases such as CHF and the Fontan procedure is
generally considered to be increased hydrostatic
pressure from venous hypertension which, at least
in part, results in loss of protein into the GI tract.
1
Interestingly, these patients do not have signicantly
elevated venous hypertension relative to similar patients
without PLE making the exact pathophysiologic process
less clear.
1
Some of these patients seem to respond to
treatments based at the level of the mucosal membrane,
implying that perhaps mucosal injury is again a primary
root cause. Some hypothesize that hemodynamic
changes associated with the Fontan procedure result
in increased mesenteric vascular resistance as a
compensatory mechanism to poor cardiac output.
17
This
may in turn damage the mucosal epithelium, increase
permeability and engorge intestinal lymphatics with
an appearance histologically similar to congenital
intestinal lymphangiectasia. There are also data to
suggest that patients with univentricular-type congenital
heart disease may have increased protein loss in the
GI tract prior to the Fontan procedure; this may reect
either a response to the initial circulatory dysfunction
of the congenital heart disease or concurrent congenital
malformation of some component of the GI tract itself.
18
Others have attempted to strengthen the argument for
mucosal injury by demonstrating that patients who have
undergone a Fontan procedure typically have elevated
inammatory markers.
19
The patient was placed on prednisone for suspected
eosinophilic gastroenteritis with rapid normalization of
her serum albumin and eosinophils and more gradual
normalization of the A1AT clearance. With weaning of
the prednisone, an increase in peripheral eosinophils
and decrease in albumin occurred prompting initiation
of oral cromolyn and budesonide. Thereafter, she
was able to eliminate prednisone use. Repeat upper
(continued on page 30)
(continued from page 25)

30 PRACTICAL GASTROENTEROLOGY • APRIL 2017
NUTRITION ISSUES IN GASTROENTEROLOGY, SERIES #162
Protein Losing Enteropathy: Diagnosis and Management
a measurable response. Nutritional strategies focusing
on protein deciency are important. A high protein
diet is recommended in patients with PLE and may
require signicantly greater protein intake (2.0-3.0 g/
kg/day) than normal (0.6-0.8 g/kg/day) to remain in a
positive nitrogen balance.
6
In patients with associated
fat malabsorption, primary or secondary intestinal
lymphangiectasia or other lymphatic disorders causing
PLE, a lowering of fat intake may decrease pressure on
the lymphatics and limit protein leakage.
15
To replace
these lost fats, medium-chain triglycerides can be tried
as these provide a source of energy rich fats and are
absorbed largely via the portal vein rather than the
lymphatics.
15
However if a very low fat diet is used > 3
weeks, a source of essential fatty acids will be necessary
and fat soluble vitamins may need to be monitored.
21
If oral intake is inadequate, enteral feedings should be
considered. If fat malabsorption has been demonstrated
based on a quantitative fecal fat collection with fat
ingested or infused enterally, then a semi-elemental or
elemental product should be used. If the patient fails
enteral, then parenteral will be necessary.
Although dietary modication may not produce
obvious benet in terms of symptoms or degree of
protein wasting, the optimization of the PLE patient’s
nutritional status is important to the success of other
therapies and the patient’s overall outcome.
endoscopy approximately one year later was normal
including duodenal and gastric biopsies. Interestingly,
biopsies from the upper esophagus demonstrated
marked eosinophilia consistent with eosinophilic
esophagitis. Notably, she denied dysphagia or any other
esophageal symptoms.
Management of PLE
Because PLE is a rare disease with a variety of
seemingly disparate causes, there are limited data on
its optimal treatment. As such, no single treatment
reliably improves PLE in all patients. A core principle
is to treat the underlying disease which, if successful,
should generally result in improvement in the PLE.
Fortunately, most causes of PLE can be readily diagnosed
and treated. Examples might include optimization of
the management of eosinophilic gastroenteritis as
demonstrated in our case illustration, or fenestration
of the Fontan heart to improve cardiac output.
20
A number of PLE-specic strategies have been
described and include dietary, pharmacological or
surgical interventions. No controlled studies, however,
have been performed to demonstrate the utility of these
approaches. It is also important to recognize that there
is often a substantial delay in clinical response to
treatment of PLE, which may take months to display
(continued from page 28)
Table 2. Testing in PLE
¨ Test of Choice: A1AT clearance
o >27 mL/day reflects a general state of GI protein loss ~ 80% sensitive
o Diarrhea results in obligate A1AT loss, use a higher threshold (>56 mL/day)
o A1AT is sensitive to acid degradation; use acid suppression in hypersecretory state
o Spot A1AT testing of stool specimen is less sensitive and not recommended for diagnosis
o A random stool A1AT level with serum A1AT level may be a convenient surveillance
method for known PLE patients under treatment or in remission
¨ Alternatives to A1AT clearance:
o May be useful when there is a high clinical suspicion in the context of a negative A1AT
clearance given its higher sensitivity
o Technetium 99m-labelled human serum albumin (HSA) scintigraphy
§Involves radiation
No prolonged stool collection
o
51
Cr-albumin clearance
§Involves radiation
Requires collection of stool for a minimum of 4 days

NUTRITION ISSUES IN GASTROENTEROLOGY, SERIES #162
PRACTICAL GASTROENTEROLOGY • APRIL 2017 31
Protein Losing Enteropathy: Diagnosis and Management
Table 3. Etiologies of PLE
Erosive Gastrointestinal Disease
· Inflammatory bowel disease – primarily Crohn’s
23
· NSAID enteropathy
14
· Gut malignancies
· Graft vs Host disease
· Sarcoidosis
24
· Ulcerative jejunoileitis
Nonerosive Gastrointestinal Disease
· Collagenous Colitis
25
· Amyloidosis
26
· Menetrier’s
27
· Gastric Polyposis
28
· Celiac sprue
· Tropical Sprue
· Eosinophilic gastroenteritis
29
· Lymphocytic gastritis
· Bacterial infections (small bowel bacterial overgrowth, strongyloides,
14
tuberculosis,
Helicobacter pylori,
30
Whipple’s disease
31
)
· CMV with hypertrophic gastropathy
32
· Lupus & ANCA (+) vasculitis
16
· Connective tissue disorders (e.g., Sjogren’s syndrome)
· Congenital metabolic abnormalities (e.g., Gaucher’s
33
)
Lymphatic Congestion/Obstruction
· Primary lymphangiectasia
34
· Secondary lymphangiectasia (heart failure, mesenteric panniculitis, retroperitoneal fibrosis,
tuberculous infiltration, lymphoma, neoplastic lymphoid invasion)
15,35,36
· Thoracic duct obstruction
· Portal hypertension
· Congenital malformation of the lymphatics
Cardiac isease
· Congenital heart disease
· Fontan operation for univentricular heart
· Congestive heart failure (particularly right heart failure)
· Constrictive pericarditis

32 PRACTICAL GASTROENTEROLOGY • APRIL 2017
NUTRITION ISSUES IN GASTROENTEROLOGY, SERIES #162
Protein Losing Enteropathy: Diagnosis and Management
NUTRITION ISSUES IN GASTROENTEROLOGY, SERIES #162
Direct replacement of serum albumin by infusion
is not a useful long-term strategy as it provides only
short-term benet, is expensive, and does not reverse
the underlying pathophysiology. In the acute setting,
albumin infusion may help patients suffering from severe
third-spacing of uid due to marked hypoalbuminemia
as a bridge to more durable therapies.
22
Supportive measures to avoid complications
resulting from uid retention contribute meaningfully
to PLE patients’ quality of life. Use of compression
stockings may help decrease edema and improve
functional status. Careful skin care along with edema
management is important to avoid pressure ulcerations
and other complications of skin breakdown.
Although there are a number of anecdotal case
reports and small case series of medical therapies for
patients with specic causes of PLE, there are no high
quality randomized controlled trials of any therapy in
PLE (see Table 4). In some cases, a surgical approach
to the primary underlying GI pathology is necessary.
In inammatory bowel disease, for example, this
might result in resection of active bowel affected.
Gastrectomy may prove curative for patients with
Ménétrier’s disease.
Practical approach to PLE
Given the rarity of the PLE and the lack of rigorous
Table 4. Reported Treatments for PLE
Therapy
Diuretics · Considered unhelpful with managing the anasarca
· May be considered in those with cardiac causes
High-dose
Spironolactone
· Hypothesized to have an effect on the physiology of PLE beyond
simply acting as a diuretic, perhaps through similar pleiotropic
mechanisms that bring about its benefit in CHF and cirrhosis
37
Heparin · May stabilize capillary or mucosal endothelium
· Retrospective review of 17 patients suggesting improved
symptoms but not objective outcomes
38,39
Corticosteroids · Case reported data in Fontan-associated PLE suggesting benefit
40
Budesonide · 4 small cases series (total 31 patients w/ PLE after Fontan)
suggesting clinical and biochemical improvement with
9mg/day dosing
19,41-43
Octreotide · Case reported in treatment for PLE from a variety of causes
· May favorably modify intestinal blood or lymphatic flow and
decrease vascular permeability through modulation of
vasoactive peptides
13,26,44,45
Cetuximab* · Case reported in treatment for PLE associated with Ménétrier’s
46
Everolimus** · Case reported in treatment for PLE associated with primary
intestinal lymphangiectasia
47
* Epidermal growth factor receptor monoclonal antibody
**mTOR kinase Inhibitor
(continued on page 34)

34 PRACTICAL GASTROENTEROLOGY • APRIL 2017
NUTRITION ISSUES IN GASTROENTEROLOGY, SERIES #162
Protein Losing Enteropathy: Diagnosis and Management
supportive data, the treatment of PLE can be a puzzle
to the clinician. The initial step in the evaluation of
hypoalbuminemia is to exclude other, more common,
causes such as liver and renal diseases. When
concern over PLE remains, the A1AT clearance
test is recommended as the test of choice given its
reliability, relative inexpense, and wide availability.
After a diagnosis of PLE has been made, the following
approach is suggested:
1. Aggressive pursuit to identify the underlying
disease responsible for PLE and treat accordingly
while encouraging a high protein diet and
supportive measures for uid retention when
present. A low-fat diet and supplementation with
medium chain triglyceride supplementation
should be considered on an individual basis.
Monitoring and treatment of any associated
malnutrition and/or micronutrient deciencies,
when present, is also important. In certain
conditions where severe symptoms prevent
adequate oral intake, occasionally use of either
enteral or parenteral support may be needed.
2.
If cardiac disease is an underlying etiology,
addition of diuretic regimen including
spironolactone should be considered. The
chronic use of diuretics in other settings,
while commonly attempted, is otherwise
generally discouraged as is the long-term use
of intermittent albumin infusions. The use of
other “heart failure” medications in order to
optimize cardiac output in those with cardiac
etiologies of PLE may also be pursued.
3.
A trial of pharmacological agents such as
corticosteroids, heparin, and octreotide
may be considered in patients who have not
responded to other measures, but should not
be considered primary therapeutic agents for
PLE. The use of cetuximab and everolimus
should be considered based upon its reported
use in relevant underlying diseases. If a trial
of budesonide 9mg daily results in clinical
improvement, a gradual taper over several
months is recommended, recognizing that
recurrence is common after discontinuation.
Subcutaneous heparin or octreotide may be
used in combination with oral or intravenous
corticosteroids.
4.
Periodic monitoring of the degree of PLE
is advised, for example, by using A1AT
clearance, after treatment is initiated. Similarly,
periodically monitoring the serum albumin
level is recommended with the assessment of
other serum chemistries and micronutrients on
a case-by-case basis.
CONCLUSION
The patient described in our case illustration represents
a typical case of PLE with a GI etiology with the
exception of an absence of edema. It highlights the
need to recognize PLE as a cause of unexplained
hypoalbuminemia even in the absence of GI symptoms
or evidence of uid retention. PLE can occur in the
context of a myriad of diseases ranging from primary
GI mucosal disorders, to malignancies, to lymphatic
disorders, to congenital heart disease. Diagnosis is most
commonly conrmed by the A1AT clearance test. The
care of the patient with PLE can be challenging and
often requires a multidisciplinary approach. While the
evidence regarding the management of PLE is limited,
treatment primarily focuses of the underlying disease
with the addition of supportive measures to manage
complications such as edema.
References
1. Rychik J. Protein-losing enteropathy after Fontan operation.
Congenital heart disease. 2007;2(5):288-300.
2. Chng HH et al. Major gastrointestinal manifestations in
lupus patients in Asia: lupus enteritis, intestinal pseudo-
obstruction, and protein-losing gastroenteropathy. Lupus.
2010;19(12):1404-1413.
3. Biancone L, Fantini M, Tosti C, et al. Fecal alpha 1-anti-
trypsin clearance as a marker of clinical relapse in patients
with Crohn’s disease of the distal ileum. Eur J Gastroenterol
Hepatol. 2003;15(3):261-266.
4. Eustace PW, Gaunt JI, Croft DN. Incidence of protein-losing
enteropathy in primary lymphoedema using chromium-51
chloride technique. Br Med J. 1975;4(5999):737.
5. Schmidt PN, Blirup-Jensen S, Svendsen PJ, et al.
Characterization and quantication of plasma proteins
excreted in faeces from healthy humans. Scand J Clin Lab
Invest. 1995;55(1):35-45.
6. Umar SB, DiBaise JK. Protein-losing enteropathy: case
illustrations and clinical review. Am J Gastroenterol.
2010;105(1):43-49; quiz 50.
7. Levitt DG, Levitt MD. Human serum albumin homeostasis: a
new look at the roles of synthesis, catabolism, renal and gas-
trointestinal excretion, and the clinical value of serum albumin
measurements. Int J Gen Med. 2016;9:229-255.
8. Takeda H, Ishihama K, Fukui T, et al. Signicance of
rapid turnover proteins in protein-losing gastroenteropathy.
Hepatogastroenterology. 2003;50(54):1963-1965.
(continued from page 32)

NUTRITION ISSUES IN GASTROENTEROLOGY, SERIES #162
PRACTICAL GASTROENTEROLOGY • APRIL 2017 35
Protein Losing Enteropathy: Diagnosis and Management
9. Udink Ten Cate FE, Hannes T, Germund I, et al. Towards a
proposal for a universal diagnostic denition of protein-losing
enteropathy in Fontan patients: a systematic review. Heart
(British Cardiac Society). 2016;102(14):1115-1119.
10. Strygler B, Nicar MJ, Santangelo WC, et al. Alpha 1-antitryp-
sin excretion in stool in normal subjects and in patients with
gastrointestinal disorders. Gastroenterology. 1990;99(5):1380-
1387.
11. Takeda H, Nishise S, Furukawa M, et al. Fecal clear-
ance of alpha1-antitrypsin with lansoprazole can detect pro-
tein-losing gastropathy. Digestive diseases and sciences.
1999;44(11):2313-2318.
12. Chau TN, Mok MY, Chan EY, et al. Evaluation of perfor-
mance of measurement of faecal alpha(1)-antitrypsin clear-
ance and technetium-99m human serum albumin scintigraphy
in protein-losing enteropathy. Digestion. 2011;84(3):199-206.
13. John AS, Johnson JA, Khan M, et al. Clinical outcomes and
improved survival in patients with protein-losing enteropathy
after the Fontan operation. J Am Coll Cardiol. 2014;64(1):54-
62.
14. Takenaka H, Ohmiya N, Hirooka Y, et al. Endoscopic
and imaging ndings in protein-losing enteropathy. J Clin
Gastroenterol. 2012;46(7):575-580.
15. Tift WL, Lloyd JK. Intestinal lymphangiectasia. Long-term
results with MCT diet. Arch Dis Child. 1975;50(4):269-276.
16. Law ST, Ma KM, Li KK. Protein-losing enteropathy associ-
ated with or without systemic autoimmune disease: what are
the differences? Eur J Gastroenterol Hepatol. 2012;24(3):294-
302.
17. Ostrow AM, Freeze H, Rychik J. Protein-losing enteropathy
after fontan operation: investigations into possible pathophysi-
ologic mechanisms. Ann Thorac Surg. 2006;82(2):695-700.
18. Patel JK, Loomes KM, Goldberg DJ, et al. Early Impact of
Fontan Operation on Enteric Protein Loss. Ann Thorac Surg.
2016;101(3):1025-1030.
19. Thacker D, Patel A, Dodds K, et al. Use of oral budesonide in
the management of protein-losing enteropathy after the Fontan
operation. Ann Thorac Surg. 2010;89(3):837-842.
20. Jacobs ML, Rychik J, Byrum CJ, et al. Protein-losing enter-
opathy after Fontan operation: resolution after bafe fenestra-
tion. Ann Thorac Surg. 1996;61(1):206-208.
21. Shah ND LB. The Use of Medium-Chain Triglycerides in
Gastrointestinal Disorders. Practical Gastroenterology. 2017.
[In Press]
22. Rychik J, Spray TL. Strategies to treat protein-losing enteropa-
thy. Semin Thorac Cardiovasc Surg Pediatr Card Surg Annu.
2002;5:3-11.
23. Baert D, Wulfrank D, Burvenich P, et al. Lymph loss in the
bowel and severe nutritional disturbances in Crohn’s disease.
J Clin Gastroenterol. 1999;29(3):277-279.
24. Popovic OS, Brkic S, Bojic P, et al. Sarcoidosis and protein
losing enteropathy. Gastroenterology. 1980;78(1):119-125.
25. Nakaya Y, Hosokawa SK, Kataoka Y, et al. Collagenous
Colitis Associated with Protein-losing Enteropathy. Intern
Med. 2015;54(22):2959-2960.
26. Fushimi T, Takahashi Y, Kashima Y, et al. Severe protein
losing enteropathy with intractable diarrhea due to systemic
AA amyloidosis, successfully treated with corticosteroid and
octreotide. Amyloid. 2005;12(1):48-53.
27. Meuwissen SG, Ridwan BU, Hasper HJ, et al. Hypertrophic
protein-losing gastropathy. A retrospective analysis of 40 cases
in The Netherlands. The Dutch Menetrier Study Group. Scand
J Gastroenterol Suppl. 1992;194:1-7.
28. Yamashita K, Saito M, Itoh M, et al. Juvenile polyposis
complicated with protein losing gastropathy. Intern Med.
2009;48(5):335-338.
29. Siewert E, Lammert F, Koppitz P, et al. Eosinophilic gastro-
enteritis with severe protein-losing enteropathy: successful
treatment with budesonide. Dig Liver Dis. 2006;38(1):55-59.
30. Cohen HA, Shapiro RP, Frydman M, et al. Childhood protein-
losing enteropathy associated with Helicobacter pylori infec-
tion. J Pediatr Gastroenterol Nutr. 1991;13(2):201-203.
31. Freeman HJ. Tropheryma whipplei infection. World J
Gastroenterol. 2009;15(17):2078-2080.
32. Megged O, Schlesinger Y. Cytomegalovirus-associated
protein-losing gastropathy in childhood. Eur J Pediatr.
2008;167(11):1217-1220.
33. Mhanni AA, Kozenko M, Hartley JN, et al. Successful therapy
for protein-losing enteropathy caused by chronic neurono-
pathic Gaucher disease. Mol Genet Metab Rep. 2016;6:13-15.
34. Vignes S, Bellanger J. Primary intestinal lymphangiectasia
(Waldmann’s disease). Orphanet J Rare Dis. 2008;3:5.
35. Tsukamoto A, Nakamura F, Nannya Y, et al. MALT lymphoma
of the small bowel with protein-losing enteropathy. Int J
Hematol. 2014;99(2):198-201.
36. Pratz KW, Dingli D, Smyrk TC, et al. Intestinal lymphangiec-
tasia with protein-losing enteropathy in Waldenstrom macro-
globulinemia. Medicine (Baltimore). 2007;86(4):210-214.
37. Ringel RE, Peddy SB. Effect of high-dose spironolactone
on protein-losing enteropathy in patients with Fontan pal-
liation of complex congenital heart disease. Am J Cardiol.
2003;91(8):1031-1032, A1039.
38. Donnelly JP, Rosenthal A, Castle VP, et al. Reversal of pro-
tein-losing enteropathy with heparin therapy in three patients
with univentricular hearts and Fontan palliation. J Pediatr.
1997;130(3):474-478.
39. Ryerson L, Goldberg C, Rosenthal A, et al. Usefulness of
heparin therapy in protein-losing enteropathy associated with
single ventricle palliation. Am J Cardiol. 2008;101(2):248-
251.
40. Rychik J, Piccoli DA, Barber G. Usefulness of corticosteroid
therapy for protein-losing enteropathy after the Fontan proce-
dure. Am J Cardiol. 1991;68(8):819-821.
41. Turner Z, Lanford L, Webber S. Oral budesonide as a therapy
for protein-losing enteropathy in patients having undergone
Fontan palliation. Congenital heart disease. 2012;7(1):24-30.
42. Gursu HA, Erdogan I, Varan B, et al. Oral budesonide as a
therapy for protein-losing enteropathy in children after the
Fontan operation. J Card Surg. 2014;29(5):712-716.
43. Schumacher KR, Cools M, Goldstein BH, et al. Oral
budesonide treatment for protein-losing enteropathy in Fontan-
palliated patients. Pediatr Cardiol. 2011;32(7):966-971.
44. Filik L, Oguz P, Koksal A, et al. A case with intestinal lym-
phangiectasia successfully treated with slow-release octreo-
tide. Dig Liver Dis. 2004;36(10):687-690.
45. Rothenberg M, Pai R, Stuart K. Successful use of octreotide
to treat Menetrier’s disease: a rare cause of abdominal pain,
weight loss, edema, and hypoalbuminemia. Digestive diseases
and sciences. 2009;54(7):1403-1407.
46. Settle SH, Washington K, Lind C, et al. Chronic treatment
of Menetrier’s disease with Erbitux: clinical efcacy and
insight into pathophysiology. Clin Gastroenterol Hepatol.
2005;3(7):654-659.
47. Ozeki M, Hori T, Kanda K, et al. Everolimus for Primary
Intestinal Lymphangiectasia With Protein-Losing Enteropathy.
Pediatrics. 2016;137(3):e20152562.
